Ehlers-Danlos syndrome
nosology by P. Tsipouras, M.D.
adapted by prof nivaldo baldo
ABSTRACT
The categorization of the Ehlers-Danlos syndromes began in the late 1960's and was formalized in the Berlin nosology. Over time, it became apparent that the diagnostic criteria established and published in 1988 did not discriminate adequately between the different types of the Ehlers-Danlos syndrome or between the Ehlers-Danlos syndrome and other phenotypically related conditions. In addition, the elucidation of the molecular basis of several Ehlers-Danlos syndromes has added a new dimension to the characterization of this group of disorders. We propose a revision of the classification of the Ehlers-Danlos syndromes based primarily on the cause of each type. Major and minor diagnostic criteria have been defined for each type and complemented whenever possible with laboratory findings. This simplified classification will facilitate an accurate diagnosis of the Ehlers-Danlos syndromes and contribute to the delineation of phenotypically related disorders.
Key Words:
nosology by P. Tsipouras, M.D.
adapted by prof nivaldo baldo
ABSTRACT
The categorization of the Ehlers-Danlos syndromes began in the late 1960's and was formalized in the Berlin nosology. Over time, it became apparent that the diagnostic criteria established and published in 1988 did not discriminate adequately between the different types of the Ehlers-Danlos syndrome or between the Ehlers-Danlos syndrome and other phenotypically related conditions. In addition, the elucidation of the molecular basis of several Ehlers-Danlos syndromes has added a new dimension to the characterization of this group of disorders. We propose a revision of the classification of the Ehlers-Danlos syndromes based primarily on the cause of each type. Major and minor diagnostic criteria have been defined for each type and complemented whenever possible with laboratory findings. This simplified classification will facilitate an accurate diagnosis of the Ehlers-Danlos syndromes and contribute to the delineation of phenotypically related disorders.
Key Words:
Ehlers-Danlos syndromes, diagnosis, joint hypermobility, skin extensibility, tissue fragility, arterial rupture, heritable disorders of connective tissue.
INTRODUCTION
The Ehlers-Danlos syndromes (EDS) are a heterogeneous group of heritable connective tissue disorders characterized by articular hypermobility, skin extensibility and tissue fragility. Classification of the EDS began in the late 1960's [Beighton, 1970, McKusick, 1972] and in 1986, a nosology was proposed at a meeting in Berlin , which formalized the nomenclature of the various types [Beighton et al., 1988]. Recent developments in the elucidation of the biochemical and molecular basis of the EDS, together with increasing clinical experience, now permit refinement of the existing nosology. We met in June 1997 at Villefranche-sur-Mer, , in order to discuss the revision of the classification. Our proposals, which form the subject of this communication, have the aim of facilitating development and improvement in the following aspects of the EDS: 1) diagnostic uniformity for clinical and research purposes, 2) natural history, 3) management, 4) genetic counseling, 5) identification of potential areas of research. We propose a new simplified classification of the EDS into 6 major types. Our guiding principle in formulating the proposed classification was its usefulness to the "generalist". For each type, we defined major and minor diagnostic criteria. A major criterion has high diagnostic specificity, because it is infrequent in other conditions and in the general population. The presence of one or more major criteria is either necessary for clinical diagnosis or highly indicative and warrants laboratory confirmation whenever possible. A minor criterion is a sign of lesser diagnostic specificity. The presence of one or more minor criteria contributes to the diagnosis of a specific type of EDS, however in the absence of major criteria they are not sufficient to establish the diagnosis. The presence of minor criteria might be suggestive of the diagnosis of (an) EDS-like condition(s), the nature of which will be elucidated when the molecular basis becomes known.
METHODS The authors arrived at the proposed classification through a review of known clinical data and the biochemical and molecular observations obtained since the Berlin nosology meeting. This document was then circulated to other professionals working in the field for review and criticism.
REVISED CLASSIFICATION
A) General Comments
1. Skin hyperextensibility should be tested at a neutral site, meaning a site not subjected to mechanical forces or scarring, e.g., the volar surface of the forearm. It is measured by pulling up the skin until resistance is felt. In young children, it is difficult to assess because of the abundance of subcutaneous fat [Beighton, 1993; Steinmann et al., 1993].
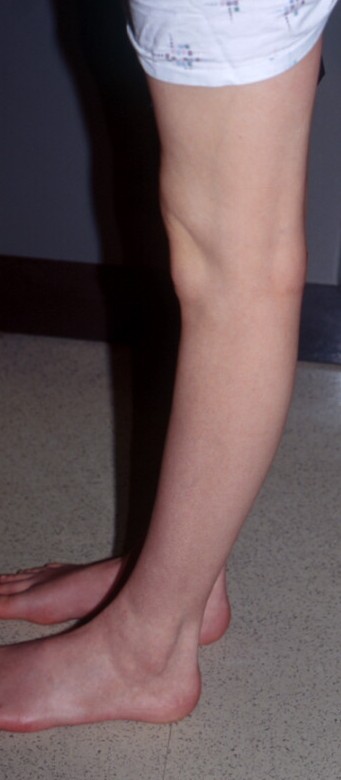
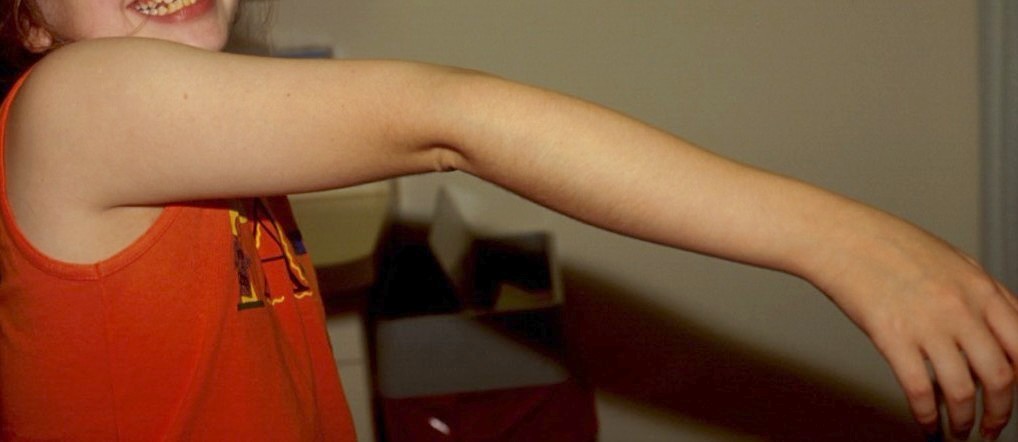
2. Joint hypermobility should be assessed using the Beighton scale [Beighton et al., 1983]. Joint hypermobility depends on age, gender, family, and ethnic background. A score of 5/9 or greater defines hypermobility. The total score is obtained by: a) passive dorsiflexion of the little fingers beyond 90 degrees; one point for each hand. b) passive apposition of the thumbs to the flexor aspect of the forearm; one point for each hand. c) Hyperextension of the elbows beyond 10 degrees; one point for each elbow. d) Hyperextension of the knees beyond 10 degrees; one point for each knee. e) forward flexion of the trunk with knees fully extended so that the palms of the hand rest flat on the floor; one point. Easy bruising manifests as spontaneous ecchymoses, frequently recurring in the same areas, and causing characteristic brownish discoloration. Easy bruising may be the presenting symptom in early childhood. Child abuse should be considered in the differential diagnosis. There is tendency to prolonged bleeding in spite of normal coagulation status [Beighton, 1993; Steinmann et al., 1993].
4. Tissue fragility manifests as easy bruising and the presence of dystrophic scars. Scars are found mostly on pressure points (knee, elbow, forehead, chin) and have a thin, atrophic papyraceous appearance. Frequently the scars become wide and discolored; wound healing is impaired [Beighton, 1993; Steinmann et al., 1993].
5. Mitral valve prolapse (MVP) and proximal aortic dilatation should be diagnosed by echocardiography, CT or MRI. Mitral valve prolapse is a common manifestation, but aortic dilatation is uncommon; in a small proportion of patients with EDS it may be progressive [Leier et al., 1980]. Dilatation of the aortic root should be diagnosed when the maximum diameter at the sinuses of Valsalva exceeds the upper normal limits for age and body size [Roman et al., 1989, Roman et al., 1993]. Stringent criteria should be used for diagnosis of MVP [Devereux et al., 1987]. In those individuals where aortic dilatation exists, anuloaortic ectasia needs to be considered in the differential diagnosis.
6. Chronic joint and limb pain is common and skeletal radiographs are normal [Sacheti et al., 1997]. Frequently it is difficult to establish the precise anatomical localization of the pain.
7. Although well defined, the Kyphoscoliosis, Arthrochalasia, and Dermatosparaxis Types are considerably less common than the Classical, Hypermobility, and Arterial Types [Beighton, 1993; Steinmann et al., 1993].
B) Classification
1) Classical Type
i) Inheritance
*autosomal dominant
ii) Major diagnostic criteria
*skin hyperextensibility
*widened atrophic scars--(manifestation of tissue fragility)
*joint hypermobility
iii) Minor diagnostic criteria
*smooth velvety skin
*molluscoid pseudotumors
*subcutaneous spheroids
*complications of joint hypermobility (sprains, dislocations/subluxations, pes planus, etc.; Beighton and Horan, 1969)
*muscle hypotonia, delayed gross motor development
*easy bruising
*manifestations of tissue extensibility and fragility (e.g., hiatal hernia, anal prolapse in childhood, cervical insufficiency) [Steinmann et al., 1993]
*surgical complications (post-operative hernias); [Beighton and Horan, 1960, Steinmann et al., 1993]
*positive family history
iv) Cause and laboratory diagnosis:
Abnormal electrophoretic mobility of the proa1(V) or proa2(V) chains of collagen type V has been detected in several but not all families with the Classical Type. Because a highly sensitive screening method has not yet been developed, the absence of detected abnormalities by biochemical or molecular analysis does not rule out a defect in collagen type V. In informative families genetic linkage studies can be used for prenatal and postnatal diagnosis. Mutation analysis in individuals is being performed on a research basis. Locus heterogeneity has been documented (Steinmann et al., 1993). Genetic linkage to intragenic markers of the COL5A1 or COL5A2 genes has been excluded in some families. Abnormalities in the collagen fibril structure can be found in many families by electron microscopy [Vogel et al., 1979]; a "cauliflower" deformity of collagen fibrils, is characteristic [Hausser and Anton-Lamprecht, 1994] but not specific.
v) Special comments
1. The skin manifestations range in severity; families with mild, moderate and severe expression have been described (Table)
2. Molluscoid pseudotumors are fleshy lesions associated with scars. They are frequently found over pressure points (e.g., elbows).
3. Spheroids are small subcutaneous spherical hard bodies, frequently mobile and palpable on the forearms and shins. Spheroids may be calcified and detectable radiologically.
4. Recurrent joint subluxations are frequent in the shoulder, patella, and temporomandibular joints.
5. Dyspareunia and sexual dysfunction are occasional complaints in the Classical and other types of EDS [Sorokin et al., 1994].
6. Fatigue is a frequent complaint.
7. For management, see Steinmann et al. 1993.
2) Hypermobility Type
i) Inheritance
*autosomal dominant
ii) Major diagnostic criteria
*Skin involvement (hyperextensibility and/or smooth, velvety skin)
*Generalized joint hypermobility
iii) Minor diagnostic criteria
*recurring joint dislocations
*chronic joint/limb pain
*positive family history
iv) Special comments
1. Skin extensibility is variable. The presence of atrophic scars in individuals with joint hypermobility suggests the diagnosis of the Classical Type.
2. Joint hypermobility is the dominant clinical manifestation. Certain joints, such as the shoulder, patella, and temporo-mandibular joints dislocate frequently.
3. In rheumatologic practice, large numbers of patients present with generalized joint hypermobility [Beighton et al., 1983]. It is important to distinguish these individuals from those affected with the Hypermobility Type of EDS. There is considerable debate as to the causal interrelationships, if any, between the phenotype in such persons, and that of the Hypermobility Type of EDS.
4. Musculoskeletal pain is early in onset, chronic, and may be debilitating (Sacheti et al., 1997). The anatomical distribution is wide and tender points can sometimes be elicited. A tender point is defined as an area that, when palpated with the thumb or 2 or 3 fingers will be painful at a pressure of 4 kg or less [Wolfe et al., 1990].
5. For management, see Steinmann et al., 1993.
3) Vascular Type
Caused by structural defects in the proa1(III) chain of collagen type III encoded by COL3A1.
i) Inheritance
*autosomal dominant
ii) Major diagnostic criteria
*Thin translucent skin
*Arterial/intestinal/uterine fragility or rupture
*Extensive bruising
*Characteristic facial appearance
iii) Minor diagnostic criteria
*Acrogeria
*Hypermobility of small joints
*Tendon and muscle rupture
*Talipes equinovarus (clubfoot)
*Early onset varicose veins
*Arteriovenous, carotid-cavernous sinus fistula
*Pneumothorax/pneumohemothorax
*Gingival recession
*Positive family history, sudden death in (a) close relative(s)
The presence of any two or more of the major criteria is highly indicative of the diagnosis and laboratory testing is strongly recommended.
iv) Cause and laboratory diagnosis:
The method of laboratory diagnosis involves:
1) the demonstration of structurally abnormal collagen type III produced by fibroblasts causing defective secretion, post-translational overmodification, thermal instability, and/or sensitivity to proteases,
2) the demonstration of a mutation in the COL3A1 gene [Steinmann et al., 1993].
Determination of the serum level of procollagen type III aminopropeptide is experimental because of biological variability, confounding concomitant conditions, and analytical modification of the assay necessary for the detection of low levels [Steinmann et al., 1989].
v) Specific comments
1. Facial appearance is characteristic in some affected individuals ( Fig 1). There is a decrease in the subcutaneous adipose tissue, particularly in the face and limbs.
2. Joint hypermobility is usually limited to the digits.
3. Spontaneous arterial rupture has a peak incidence in the third or fourth decade of life but may occur earlier. Midsize arteries are most commonly involved. Arterial rupture is the most common cause of sudden death [Pepin et al., 1992].
4. Acute abdominal and flank pain (diffuse or localized) is a common presentation of arterial or intestinal rupture and should be investigated urgently. Non-invasive diagnostic procedures are recommended.
5. The subcutaneous venous pattern is particularly apparent over the chest and abdomen.
6. In the presence of severe bruising as an initial complication, child abuse and/or hematological disorders need to be considered. In the context of chronic bruising and abnormal scar formation, differentiation from the Classical Type of EDS is necessary.
7. Diagnosis of this condition is difficult in children in the absence of a family history.
8. Pregnancies may be complicated by intra-partum uterine rupture and pre- and post-partum arterial bleeding. Vaginal and perineal tears may be sustained during delivery.
9. Complications during and after surgery (e.g., wound dehiscence) are frequent and severe.
10. For management, see Steinmann et al., 1993.
4) Kyphoscoliosis Type
Caused by a deficiency of lysyl hydroxylase (PLOD), a collagen-modifying enzyme. Homozygosity or compound heterozygosity for mutant PLOD allele(s) results in the deficiency.
i) Inheritance
*autosomal recessive
ii) Major diagnostic criteria
*Generalized joint laxity
*Severe muscle hypotonia at birth
*Scoliosis at birth, progressive
*Scleral fragility and rupture of the ocular globe
iii) Minor diagnostic criteria
*Tissue fragility, including atrophic scars
*Easy bruising
*Arterial rupture
*Marfanoid habitus
*Microcornea
*Radiologically considerable osteopenia
*Family history, i.e., affected sibs
The presence of three major criteria in an infant is suggestive of the diagnosis and laboratory testing is warranted.
iv) Cause and laboratory diagnosis
The recommended laboratory test is the measurement of total urinary hydroxylysyl pyridinoline and lysyl pyridinoline cross links after hydrolysis by HPLC, a test which is readily available and has a very high degree of sensitivity and specificity [Steinmann et al., 1995]. The determination of dermal hydroxylysine is also easy; however determination of lysyl hydroxylase activity in fibroblasts and/or mutational analysis of the PLOD gene is performed on a research basis only.
v) Specific comments
1. Muscular hypotonia can be very pronounced and leads to delayed gross motor development. This condition should be considered in the initial differential diagnosis of a floppy infant [Wenstrup et al., 1989; Steinmann et al., 1993].
2. The phenotype is most often severe, frequently resulting in loss of ambulation in the second or third decade.
3. Scleral fragility may lead to rupture of the ocular globe after minor trauma. The condition should be differentiated from the brittle cornea syndrome [Royce et al., 1990]. It is now apparent that serious eye complications are much less frequent than previously thought [Wenstrup et al., 1989; Steinmann et al., 1993], hence the change in the descriptor of this type .
4. The severe neonatal form of Marfan syndrome should be considered in the differential diagnosis.
5. There have been reports of a less severe form of the condition with normal activity of lysyl hydroxylase and a normal hydroxylysine content in the dermis (OMIM # 229200); this form is even rarer.
6. For management, see Steinmann et al. [1993].
5) Arthrochalasia Type
Caused by mutations leading to deficient processing of the amino-terminal end of proa1(I) [type A] or proa2(I) [type B] chains of collagen type I because of skipping of exon 6 in either gene.
i) Inheritance
*autosomal dominant
i) Major diagnostic criteria
*Severe generalized joint hypermobility with recurrent subluxations
*Congenital bilateral hip dislocation.
iii) Minor diagnostic criteria
*Skin hyperextensibility
*Tissue fragility, including atrophic scars
*Easy bruising
*Muscle hypotonia
*Kyphoscoliosis
*Radiologically mild osteopenia
iv) Cause and laboratory diagnosis:
The biochemical defect is determined by electrophoretic demonstration of pNa1(I) or pNa2(I) chains extracted from dermal collagen or harvested from cultured skin fibroblasts. Direct demonstration of complete or partial exon 6 skipping in cDNAs of COL1A1 or COL1A2, respectively, can be performed, followed by mutation analysis [Steinmann et al., 1993].
v) Special comments
1. Congenital hip dislocation has been present in all biochemically proven individuals.
2. Short stature is not a manifestation, unless it is a complication of severe kyphoscoliosis and/or hip dislocation.
3. Larsen syndrome should be considered in the differential diagnosis.
4. For management, see Steinmann et al. [1993].
6) Dermatosparaxis Type
Caused by deficiency of procollagen I N-terminal peptidase caused by homozygosity or compound heterozygosity of mutant alleles (in contrast to the Arthrochalasia Type which is due to mutations involving the substrate sites of procollagen type I chains).
i) Inheritance
*autosomal recessive
ii) Major diagnostic criteria
*Severe skin fragility
*Sagging, redundant skin
iii) Minor diagnostic criteria
*Soft, doughy skin texture
*Easy bruising
*Premature rupture of fetal membranes
*Large hernias (umbilical, inguinal)
iv) Cause and laboratory diagnosis
Biochemical confirmation is based on the electrophoretic demonstration of pNa1(I) and pNa2(I) chains from collagen type I extracted from dermis in the presence of protease inhibitors, or obtained from fibroblasts. Determination of N-proteinase activity is performed on a research basis only.
v) Special comments
1. Skin fragility and bruising are substantial. Wound healing is not impaired and the scars are not atrophic.
2. The redundancy of the facial skin results in an appearance resembling cutis laxa; however, bruising and skin fragility are not manifestations of cutis laxa.
3. The name was taken from a similar phenotype and biochemical defect previously recognized in cattle, sheep and other animals.
4. The number of patients reported is small and the phenotypic spectrum might expand.
Other Types of the EDS
The current EDS type V (X-linked) was described in a single family [Beighton and Curtis, 1985]. The current EDS type VIII is similar to the Classical Type except that in addition it presents with periodontal friability [Stewart et al., 1977]. This is a rare type of EDS. The existence of this syndrome as an autonomous entity is uncertain.
EDS type IX was redefined previously as "occipital horn syndrome", an X-linked recessive condition allelic to Menkes syndrome (OMIM # 309400) [Beighton et al., 1988].
The current EDS type X was described in one family only [Arneson et al., 1980; for comments, see Steinmann et al., 1993].
The EDS type XI termed "familial joint hypermobility syndrome" was previously removed from the EDS classification [Beighton et al., 1988]. Its relationship to the EDS is not yet defined.
CONCLUDING REMARKS
The clinical variability and genetic heterogeneity of the Ehlers-Danlos syndrome has been long recognized. The existing classification [Beighton et al., 1988] differentiates the various types of the EDS on the basis of clinical manifestations and mode of inheritance. Although this approach is valid and useful it relies heavily on the identification and subjective interpretation of signs that are semi-quantitative, e.g., skin extensibility, joint hypermobility, tissue fragility, bruising etc. The result is frequent diagnostic confusion regarding the type of EDS and the inclusion of phenotypically similar conditions under the broad diagnosis of EDS. Since the publication of the existing classification, several reports have described the clinical findings, natural history, and molecular basis of different types of the EDS. This emerging information made apparent the somewhat artificial nature of the phenotypic boundaries between the former EDS Type I and EDS Type II. Another example is the frequent misdiagnosis of Joint Hypermobility as a type of the EDS. Thus, we re-visited the existing Berlin classification with the following objectives: 1) To refine the diagnostic definitions by introducing diagnostic criteria based on the specificity of the various clinical manifestations for each type of the EDS; 2) To formalize the use of laboratory findings, whenever possible, in the diagnostic definition of each type; 3) To simplify the existing EDS classification so that it becomes more accessible to the average generalist.
The proposed classification defines 6 major types of EDS. The descriptor captures, what is, in our opinion, the pathognomonic manifestation of each type. Furthermore, the molecular basis of each of the proposed types has either been clearly defined or is emerging. Thus, we concluded that what was formerly known as EDS Type I and EDS Type II could be merged in a single entity, the proposed Classical Type, because recent evidence indicates that they can have a common cause such as mutations in the COL5A1 or COL5A2 genes. Furthermore, the earlier differentiation was based primarily on the extent of the severity of the skin manifestations, a trait that could be attributable to a phenotype/genotype correlation, and was not necessarily a distinction based on cause.
The diagnostic criteria proposed for the Hypermobility Type will permit clear distinction from other types of EDS and also from phenotypically related disorders. We define the Vascular Type of EDS on the basis of clinical manifestations and the presence of mutations in the COL3A1 gene. Similarly, we define the Kyphoscoliosis, Arthrochalasia and Dermatosparaxis Types on the basis of clinical manifestations and the presence of particular biochemical abnormalities or molecular defects.
The former EDS Type V is a rare variant, the molecular basis of which remains unknown. The clinical characteristics of the entity currently known as EDS Type VIII remain uncertain, thus its delineation would require more clinical and molecular information.
We hope that these revised criteria can serve as a new albeit provisional standard for clinical diagnosis of the Ehlers-Danlos syndrome, for investigations of its genetic heterogeneity and phenotype-genotype correlations and clinical research on various aspects of these conditions. A further aim of this publication is to provide diagnostic criteria which will allow a clearer distinction of disorders that partially overlap with EDS and aid their clinical identification and research evaluation.
ACKNOWLEDGMENTS
The Ehlers-Danlos National Foundation (USA) and the Ehlers-Danlos Support Group (UK) sponsored this endeavor. Representatives of several national EDS groups held their own first international meeting at the same time. This concurrence promoted contacts, interaction and interchange, making it possible for involved laypersons to provide valuable input into the development of concepts concerning the EDS. The authors express their thanks to Drs. Peter Byers, William Cole, Michael Pope, Peter Royce, and Andrea Superti-Furga for reviewing and criticizing the manuscript.
INTRODUCTION
The Ehlers-Danlos syndromes (EDS) are a heterogeneous group of heritable connective tissue disorders characterized by articular hypermobility, skin extensibility and tissue fragility. Classification of the EDS began in the late 1960's [Beighton, 1970, McKusick, 1972] and in 1986, a nosology was proposed at a meeting in Berlin , which formalized the nomenclature of the various types [Beighton et al., 1988]. Recent developments in the elucidation of the biochemical and molecular basis of the EDS, together with increasing clinical experience, now permit refinement of the existing nosology. We met in June 1997 at Villefranche-sur-Mer, , in order to discuss the revision of the classification. Our proposals, which form the subject of this communication, have the aim of facilitating development and improvement in the following aspects of the EDS: 1) diagnostic uniformity for clinical and research purposes, 2) natural history, 3) management, 4) genetic counseling, 5) identification of potential areas of research. We propose a new simplified classification of the EDS into 6 major types. Our guiding principle in formulating the proposed classification was its usefulness to the "generalist". For each type, we defined major and minor diagnostic criteria. A major criterion has high diagnostic specificity, because it is infrequent in other conditions and in the general population. The presence of one or more major criteria is either necessary for clinical diagnosis or highly indicative and warrants laboratory confirmation whenever possible. A minor criterion is a sign of lesser diagnostic specificity. The presence of one or more minor criteria contributes to the diagnosis of a specific type of EDS, however in the absence of major criteria they are not sufficient to establish the diagnosis. The presence of minor criteria might be suggestive of the diagnosis of (an) EDS-like condition(s), the nature of which will be elucidated when the molecular basis becomes known.
METHODS The authors arrived at the proposed classification through a review of known clinical data and the biochemical and molecular observations obtained since the Berlin nosology meeting. This document was then circulated to other professionals working in the field for review and criticism.
REVISED CLASSIFICATION
A) General Comments
1. Skin hyperextensibility should be tested at a neutral site, meaning a site not subjected to mechanical forces or scarring, e.g., the volar surface of the forearm. It is measured by pulling up the skin until resistance is felt. In young children, it is difficult to assess because of the abundance of subcutaneous fat [Beighton, 1993; Steinmann et al., 1993].
2. Joint hypermobility should be assessed using the Beighton scale [Beighton et al., 1983]. Joint hypermobility depends on age, gender, family, and ethnic background. A score of 5/9 or greater defines hypermobility. The total score is obtained by: a) passive dorsiflexion of the little fingers beyond 90 degrees; one point for each hand. b) passive apposition of the thumbs to the flexor aspect of the forearm; one point for each hand. c) Hyperextension of the elbows beyond 10 degrees; one point for each elbow. d) Hyperextension of the knees beyond 10 degrees; one point for each knee. e) forward flexion of the trunk with knees fully extended so that the palms of the hand rest flat on the floor; one point. Easy bruising manifests as spontaneous ecchymoses, frequently recurring in the same areas, and causing characteristic brownish discoloration. Easy bruising may be the presenting symptom in early childhood. Child abuse should be considered in the differential diagnosis. There is tendency to prolonged bleeding in spite of normal coagulation status [Beighton, 1993; Steinmann et al., 1993].
4. Tissue fragility manifests as easy bruising and the presence of dystrophic scars. Scars are found mostly on pressure points (knee, elbow, forehead, chin) and have a thin, atrophic papyraceous appearance. Frequently the scars become wide and discolored; wound healing is impaired [Beighton, 1993; Steinmann et al., 1993].
5. Mitral valve prolapse (MVP) and proximal aortic dilatation should be diagnosed by echocardiography, CT or MRI. Mitral valve prolapse is a common manifestation, but aortic dilatation is uncommon; in a small proportion of patients with EDS it may be progressive [Leier et al., 1980]. Dilatation of the aortic root should be diagnosed when the maximum diameter at the sinuses of Valsalva exceeds the upper normal limits for age and body size [Roman et al., 1989, Roman et al., 1993]. Stringent criteria should be used for diagnosis of MVP [Devereux et al., 1987]. In those individuals where aortic dilatation exists, anuloaortic ectasia needs to be considered in the differential diagnosis.
6. Chronic joint and limb pain is common and skeletal radiographs are normal [Sacheti et al., 1997]. Frequently it is difficult to establish the precise anatomical localization of the pain.
7. Although well defined, the Kyphoscoliosis, Arthrochalasia, and Dermatosparaxis Types are considerably less common than the Classical, Hypermobility, and Arterial Types [Beighton, 1993; Steinmann et al., 1993].
B) Classification
1) Classical Type
i) Inheritance
*autosomal dominant
ii) Major diagnostic criteria
*skin hyperextensibility
*widened atrophic scars--(manifestation of tissue fragility)
*joint hypermobility
iii) Minor diagnostic criteria
*smooth velvety skin
*molluscoid pseudotumors
*subcutaneous spheroids
*complications of joint hypermobility (sprains, dislocations/subluxations, pes planus, etc.; Beighton and Horan, 1969)
*muscle hypotonia, delayed gross motor development
*easy bruising
*manifestations of tissue extensibility and fragility (e.g., hiatal hernia, anal prolapse in childhood, cervical insufficiency) [Steinmann et al., 1993]
*surgical complications (post-operative hernias); [Beighton and Horan, 1960, Steinmann et al., 1993]
*positive family history
iv) Cause and laboratory diagnosis:
Abnormal electrophoretic mobility of the proa1(V) or proa2(V) chains of collagen type V has been detected in several but not all families with the Classical Type. Because a highly sensitive screening method has not yet been developed, the absence of detected abnormalities by biochemical or molecular analysis does not rule out a defect in collagen type V. In informative families genetic linkage studies can be used for prenatal and postnatal diagnosis. Mutation analysis in individuals is being performed on a research basis. Locus heterogeneity has been documented (Steinmann et al., 1993). Genetic linkage to intragenic markers of the COL5A1 or COL5A2 genes has been excluded in some families. Abnormalities in the collagen fibril structure can be found in many families by electron microscopy [Vogel et al., 1979]; a "cauliflower" deformity of collagen fibrils, is characteristic [Hausser and Anton-Lamprecht, 1994] but not specific.
v) Special comments
1. The skin manifestations range in severity; families with mild, moderate and severe expression have been described (Table)
2. Molluscoid pseudotumors are fleshy lesions associated with scars. They are frequently found over pressure points (e.g., elbows).
3. Spheroids are small subcutaneous spherical hard bodies, frequently mobile and palpable on the forearms and shins. Spheroids may be calcified and detectable radiologically.
4. Recurrent joint subluxations are frequent in the shoulder, patella, and temporomandibular joints.
5. Dyspareunia and sexual dysfunction are occasional complaints in the Classical and other types of EDS [Sorokin et al., 1994].
6. Fatigue is a frequent complaint.
7. For management, see Steinmann et al. 1993.
2) Hypermobility Type
i) Inheritance
*autosomal dominant
ii) Major diagnostic criteria
*Skin involvement (hyperextensibility and/or smooth, velvety skin)
*Generalized joint hypermobility
iii) Minor diagnostic criteria
*recurring joint dislocations
*chronic joint/limb pain
*positive family history
iv) Special comments
1. Skin extensibility is variable. The presence of atrophic scars in individuals with joint hypermobility suggests the diagnosis of the Classical Type.
2. Joint hypermobility is the dominant clinical manifestation. Certain joints, such as the shoulder, patella, and temporo-mandibular joints dislocate frequently.
3. In rheumatologic practice, large numbers of patients present with generalized joint hypermobility [Beighton et al., 1983]. It is important to distinguish these individuals from those affected with the Hypermobility Type of EDS. There is considerable debate as to the causal interrelationships, if any, between the phenotype in such persons, and that of the Hypermobility Type of EDS.
4. Musculoskeletal pain is early in onset, chronic, and may be debilitating (Sacheti et al., 1997). The anatomical distribution is wide and tender points can sometimes be elicited. A tender point is defined as an area that, when palpated with the thumb or 2 or 3 fingers will be painful at a pressure of 4 kg or less [Wolfe et al., 1990].
5. For management, see Steinmann et al., 1993.
3) Vascular Type
Caused by structural defects in the proa1(III) chain of collagen type III encoded by COL3A1.
i) Inheritance
*autosomal dominant
ii) Major diagnostic criteria
*Thin translucent skin
*Arterial/intestinal/uterine fragility or rupture
*Extensive bruising
*Characteristic facial appearance
iii) Minor diagnostic criteria
*Acrogeria
*Hypermobility of small joints
*Tendon and muscle rupture
*Talipes equinovarus (clubfoot)
*Early onset varicose veins
*Arteriovenous, carotid-cavernous sinus fistula
*Pneumothorax/pneumohemothorax
*Gingival recession
*Positive family history, sudden death in (a) close relative(s)
The presence of any two or more of the major criteria is highly indicative of the diagnosis and laboratory testing is strongly recommended.
iv) Cause and laboratory diagnosis:
The method of laboratory diagnosis involves:
1) the demonstration of structurally abnormal collagen type III produced by fibroblasts causing defective secretion, post-translational overmodification, thermal instability, and/or sensitivity to proteases,
2) the demonstration of a mutation in the COL3A1 gene [Steinmann et al., 1993].
Determination of the serum level of procollagen type III aminopropeptide is experimental because of biological variability, confounding concomitant conditions, and analytical modification of the assay necessary for the detection of low levels [Steinmann et al., 1989].
v) Specific comments
1. Facial appearance is characteristic in some affected individuals ( Fig 1). There is a decrease in the subcutaneous adipose tissue, particularly in the face and limbs.
2. Joint hypermobility is usually limited to the digits.
3. Spontaneous arterial rupture has a peak incidence in the third or fourth decade of life but may occur earlier. Midsize arteries are most commonly involved. Arterial rupture is the most common cause of sudden death [Pepin et al., 1992].
4. Acute abdominal and flank pain (diffuse or localized) is a common presentation of arterial or intestinal rupture and should be investigated urgently. Non-invasive diagnostic procedures are recommended.
5. The subcutaneous venous pattern is particularly apparent over the chest and abdomen.
6. In the presence of severe bruising as an initial complication, child abuse and/or hematological disorders need to be considered. In the context of chronic bruising and abnormal scar formation, differentiation from the Classical Type of EDS is necessary.
7. Diagnosis of this condition is difficult in children in the absence of a family history.
8. Pregnancies may be complicated by intra-partum uterine rupture and pre- and post-partum arterial bleeding. Vaginal and perineal tears may be sustained during delivery.
9. Complications during and after surgery (e.g., wound dehiscence) are frequent and severe.
10. For management, see Steinmann et al., 1993.
4) Kyphoscoliosis Type
Caused by a deficiency of lysyl hydroxylase (PLOD), a collagen-modifying enzyme. Homozygosity or compound heterozygosity for mutant PLOD allele(s) results in the deficiency.
i) Inheritance
*autosomal recessive
ii) Major diagnostic criteria
*Generalized joint laxity
*Severe muscle hypotonia at birth
*Scoliosis at birth, progressive
*Scleral fragility and rupture of the ocular globe
iii) Minor diagnostic criteria
*Tissue fragility, including atrophic scars
*Easy bruising
*Arterial rupture
*Marfanoid habitus
*Microcornea
*Radiologically considerable osteopenia
*Family history, i.e., affected sibs
The presence of three major criteria in an infant is suggestive of the diagnosis and laboratory testing is warranted.
iv) Cause and laboratory diagnosis
The recommended laboratory test is the measurement of total urinary hydroxylysyl pyridinoline and lysyl pyridinoline cross links after hydrolysis by HPLC, a test which is readily available and has a very high degree of sensitivity and specificity [Steinmann et al., 1995]. The determination of dermal hydroxylysine is also easy; however determination of lysyl hydroxylase activity in fibroblasts and/or mutational analysis of the PLOD gene is performed on a research basis only.
v) Specific comments
1. Muscular hypotonia can be very pronounced and leads to delayed gross motor development. This condition should be considered in the initial differential diagnosis of a floppy infant [Wenstrup et al., 1989; Steinmann et al., 1993].
2. The phenotype is most often severe, frequently resulting in loss of ambulation in the second or third decade.
3. Scleral fragility may lead to rupture of the ocular globe after minor trauma. The condition should be differentiated from the brittle cornea syndrome [Royce et al., 1990]. It is now apparent that serious eye complications are much less frequent than previously thought [Wenstrup et al., 1989; Steinmann et al., 1993], hence the change in the descriptor of this type .
4. The severe neonatal form of Marfan syndrome should be considered in the differential diagnosis.
5. There have been reports of a less severe form of the condition with normal activity of lysyl hydroxylase and a normal hydroxylysine content in the dermis (OMIM # 229200); this form is even rarer.
6. For management, see Steinmann et al. [1993].
5) Arthrochalasia Type
Caused by mutations leading to deficient processing of the amino-terminal end of proa1(I) [type A] or proa2(I) [type B] chains of collagen type I because of skipping of exon 6 in either gene.
i) Inheritance
*autosomal dominant
i) Major diagnostic criteria
*Severe generalized joint hypermobility with recurrent subluxations
*Congenital bilateral hip dislocation.
iii) Minor diagnostic criteria
*Skin hyperextensibility
*Tissue fragility, including atrophic scars
*Easy bruising
*Muscle hypotonia
*Kyphoscoliosis
*Radiologically mild osteopenia
iv) Cause and laboratory diagnosis:
The biochemical defect is determined by electrophoretic demonstration of pNa1(I) or pNa2(I) chains extracted from dermal collagen or harvested from cultured skin fibroblasts. Direct demonstration of complete or partial exon 6 skipping in cDNAs of COL1A1 or COL1A2, respectively, can be performed, followed by mutation analysis [Steinmann et al., 1993].
v) Special comments
1. Congenital hip dislocation has been present in all biochemically proven individuals.
2. Short stature is not a manifestation, unless it is a complication of severe kyphoscoliosis and/or hip dislocation.
3. Larsen syndrome should be considered in the differential diagnosis.
4. For management, see Steinmann et al. [1993].
6) Dermatosparaxis Type
Caused by deficiency of procollagen I N-terminal peptidase caused by homozygosity or compound heterozygosity of mutant alleles (in contrast to the Arthrochalasia Type which is due to mutations involving the substrate sites of procollagen type I chains).
i) Inheritance
*autosomal recessive
ii) Major diagnostic criteria
*Severe skin fragility
*Sagging, redundant skin
iii) Minor diagnostic criteria
*Soft, doughy skin texture
*Easy bruising
*Premature rupture of fetal membranes
*Large hernias (umbilical, inguinal)
iv) Cause and laboratory diagnosis
Biochemical confirmation is based on the electrophoretic demonstration of pNa1(I) and pNa2(I) chains from collagen type I extracted from dermis in the presence of protease inhibitors, or obtained from fibroblasts. Determination of N-proteinase activity is performed on a research basis only.
v) Special comments
1. Skin fragility and bruising are substantial. Wound healing is not impaired and the scars are not atrophic.
2. The redundancy of the facial skin results in an appearance resembling cutis laxa; however, bruising and skin fragility are not manifestations of cutis laxa.
3. The name was taken from a similar phenotype and biochemical defect previously recognized in cattle, sheep and other animals.
4. The number of patients reported is small and the phenotypic spectrum might expand.
Other Types of the EDS
The current EDS type V (X-linked) was described in a single family [Beighton and Curtis, 1985]. The current EDS type VIII is similar to the Classical Type except that in addition it presents with periodontal friability [Stewart et al., 1977]. This is a rare type of EDS. The existence of this syndrome as an autonomous entity is uncertain.
EDS type IX was redefined previously as "occipital horn syndrome", an X-linked recessive condition allelic to Menkes syndrome (OMIM # 309400) [Beighton et al., 1988].
The current EDS type X was described in one family only [Arneson et al., 1980; for comments, see Steinmann et al., 1993].
The EDS type XI termed "familial joint hypermobility syndrome" was previously removed from the EDS classification [Beighton et al., 1988]. Its relationship to the EDS is not yet defined.
CONCLUDING REMARKS
The clinical variability and genetic heterogeneity of the Ehlers-Danlos syndrome has been long recognized. The existing classification [Beighton et al., 1988] differentiates the various types of the EDS on the basis of clinical manifestations and mode of inheritance. Although this approach is valid and useful it relies heavily on the identification and subjective interpretation of signs that are semi-quantitative, e.g., skin extensibility, joint hypermobility, tissue fragility, bruising etc. The result is frequent diagnostic confusion regarding the type of EDS and the inclusion of phenotypically similar conditions under the broad diagnosis of EDS. Since the publication of the existing classification, several reports have described the clinical findings, natural history, and molecular basis of different types of the EDS. This emerging information made apparent the somewhat artificial nature of the phenotypic boundaries between the former EDS Type I and EDS Type II. Another example is the frequent misdiagnosis of Joint Hypermobility as a type of the EDS. Thus, we re-visited the existing Berlin classification with the following objectives: 1) To refine the diagnostic definitions by introducing diagnostic criteria based on the specificity of the various clinical manifestations for each type of the EDS; 2) To formalize the use of laboratory findings, whenever possible, in the diagnostic definition of each type; 3) To simplify the existing EDS classification so that it becomes more accessible to the average generalist.
The proposed classification defines 6 major types of EDS. The descriptor captures, what is, in our opinion, the pathognomonic manifestation of each type. Furthermore, the molecular basis of each of the proposed types has either been clearly defined or is emerging. Thus, we concluded that what was formerly known as EDS Type I and EDS Type II could be merged in a single entity, the proposed Classical Type, because recent evidence indicates that they can have a common cause such as mutations in the COL5A1 or COL5A2 genes. Furthermore, the earlier differentiation was based primarily on the extent of the severity of the skin manifestations, a trait that could be attributable to a phenotype/genotype correlation, and was not necessarily a distinction based on cause.
The diagnostic criteria proposed for the Hypermobility Type will permit clear distinction from other types of EDS and also from phenotypically related disorders. We define the Vascular Type of EDS on the basis of clinical manifestations and the presence of mutations in the COL3A1 gene. Similarly, we define the Kyphoscoliosis, Arthrochalasia and Dermatosparaxis Types on the basis of clinical manifestations and the presence of particular biochemical abnormalities or molecular defects.
The former EDS Type V is a rare variant, the molecular basis of which remains unknown. The clinical characteristics of the entity currently known as EDS Type VIII remain uncertain, thus its delineation would require more clinical and molecular information.
We hope that these revised criteria can serve as a new albeit provisional standard for clinical diagnosis of the Ehlers-Danlos syndrome, for investigations of its genetic heterogeneity and phenotype-genotype correlations and clinical research on various aspects of these conditions. A further aim of this publication is to provide diagnostic criteria which will allow a clearer distinction of disorders that partially overlap with EDS and aid their clinical identification and research evaluation.
ACKNOWLEDGMENTS
The Ehlers-Danlos National Foundation (USA) and the Ehlers-Danlos Support Group (UK) sponsored this endeavor. Representatives of several national EDS groups held their own first international meeting at the same time. This concurrence promoted contacts, interaction and interchange, making it possible for involved laypersons to provide valuable input into the development of concepts concerning the EDS. The authors express their thanks to Drs. Peter Byers, William Cole, Michael Pope, Peter Royce, and Andrea Superti-Furga for reviewing and criticizing the manuscript.
REFERENCES
Arneson MA, Hammer Schmidt DE, Furcht LT, King RA (1980): A new form of Ehlers- Danlos syndrome. Fibronectin corrects defective platelet function. JAMA 244:144-147. Beighton P, Horan FT (1960): Surgical aspects of the Ehlers-Danlos syndrome. A survey of 100 cases. Br J Surg 56:255-259. Beighton P, Horan F (1969): Orthopaedic aspects of the Ehlers-Danlos syndrome. J Bone Joint Surg 51B:444-453. Beighton P (1970): "The Ehlers-Danlos Syndrome," William Heinemann Medical Books, London . Beighton P, Grahame R, Bird H (1983): "Hypermobility of Joints," Springer, Berlin . Beighton P, Curtis D (1985): X-linked Ehlers-Danlos syndrome type V; the next generation. Clin Genet 27:472-478. Beighton P, De Paepe A, Danks D, Finidori G, Gedde-Dahl T, Goodman R, Hall JG, Hollister DW, Horton W, McKusick VA, Opitz JM, Pope FM, Pyeritz RE, Rimoin DL, Sillence D, Spranger JW, Thompson E, Tsipouras P, Viljoen D, Winship I, Young I (1988): International nosology of heritable disorders of connective tissue, Berlin, 1986. Am J Med Genet 29:581-594. Beighton P (1993): The Ehlers-Danlos syndromes. In "Heritable Disorders of Connective Tissue," 5th Ed, pp 189-251, Mosby, St. Louis . Devereux RB, Kramer-Fox R, Shear MK, Kligfield P, Pini R, Savage DD (1987): Diagnosis and classification of severity of mitral valve prolapse: Methodologic, biologic and prognostic considerations. Am Heart J 113:1265-1280. Hausser I, Anton-Lamprecht I (1994): Differential ultrastructural aberrations of collagen fibrils in Ehlers-Danlos syndrome types I-IV as a means of diagnostics and classification. Hum Genet 3:394-407. Leier CV, Call TD, Fulkerson PK , Wooley CF (1980): The spectrum of cardiac defects in the Ehlers-Danlos syndrome, types I and III. Ann Inter Med 92(Part 1):171- 178. McKusick VA (1972): The Ehlers-Danlos syndrome. In "Heritable Disorders of Connective Tissue," 4th ed, pp 292-371, CV Mosby, St. Louis . Pepin MG, Superti-Furga A, Byers PH (1992): Natural history of Ehlers-Danlos syndrome type IV (EDS type IV): review of 137 cases. Am J Hum Genet 51:A44. Roman MJ, Devereux RB, Kramer-Fox R, O'Ranghlin J (1989): Two dimensional aortic root dimensions in normal children and adults. Am J Cardiol 64:507-512. Roman MJ, Rosen SS, Kramer-Fox R, Devereux RB (1993): The prognostic significance of the pattern of aortic root dilatation in the Marfan syndrome. J Am Coll Cardiol 22:1470-1476. Royce PM, Steinmann B, Vogel A, Steinhorst U, Kohlschuetter A (1990): Brittle cornea syndrome: A heritable connective tissue disorder distinct from Ehlers-Danlos syndrome type VI and fragilitas oculi, with spontaneous perforations of the eye, blue sclerae, red hair and normal collagen lysyl hydroxylation. Eur J Pediatr 149:465-469. Sacheti A, Szemere J, Bernstein B, Tafas T, Schechter N, Tsipouras P (1997): Chronic pain is a manifestation of the Ehlers-Danlos syndrome. J Pain Symptom Managem in press. Sorokin Y, Johnson MP, Rogowski N, Richardson DA, Evans MI (1994): Obstetric and gynecologic dysfunction in the Ehlers-Danlos syndrome.
Arneson MA, Hammer Schmidt DE, Furcht LT, King RA (1980): A new form of Ehlers- Danlos syndrome. Fibronectin corrects defective platelet function. JAMA 244:144-147. Beighton P, Horan FT (1960): Surgical aspects of the Ehlers-Danlos syndrome. A survey of 100 cases. Br J Surg 56:255-259. Beighton P, Horan F (1969): Orthopaedic aspects of the Ehlers-Danlos syndrome. J Bone Joint Surg 51B:444-453. Beighton P (1970): "The Ehlers-Danlos Syndrome," William Heinemann Medical Books, London . Beighton P, Grahame R, Bird H (1983): "Hypermobility of Joints," Springer, Berlin . Beighton P, Curtis D (1985): X-linked Ehlers-Danlos syndrome type V; the next generation. Clin Genet 27:472-478. Beighton P, De Paepe A, Danks D, Finidori G, Gedde-Dahl T, Goodman R, Hall JG, Hollister DW, Horton W, McKusick VA, Opitz JM, Pope FM, Pyeritz RE, Rimoin DL, Sillence D, Spranger JW, Thompson E, Tsipouras P, Viljoen D, Winship I, Young I (1988): International nosology of heritable disorders of connective tissue, Berlin, 1986. Am J Med Genet 29:581-594. Beighton P (1993): The Ehlers-Danlos syndromes. In "Heritable Disorders of Connective Tissue," 5th Ed, pp 189-251, Mosby, St. Louis . Devereux RB, Kramer-Fox R, Shear MK, Kligfield P, Pini R, Savage DD (1987): Diagnosis and classification of severity of mitral valve prolapse: Methodologic, biologic and prognostic considerations. Am Heart J 113:1265-1280. Hausser I, Anton-Lamprecht I (1994): Differential ultrastructural aberrations of collagen fibrils in Ehlers-Danlos syndrome types I-IV as a means of diagnostics and classification. Hum Genet 3:394-407. Leier CV, Call TD, Fulkerson PK , Wooley CF (1980): The spectrum of cardiac defects in the Ehlers-Danlos syndrome, types I and III. Ann Inter Med 92(Part 1):171- 178. McKusick VA (1972): The Ehlers-Danlos syndrome. In "Heritable Disorders of Connective Tissue," 4th ed, pp 292-371, CV Mosby, St. Louis . Pepin MG, Superti-Furga A, Byers PH (1992): Natural history of Ehlers-Danlos syndrome type IV (EDS type IV): review of 137 cases. Am J Hum Genet 51:A44. Roman MJ, Devereux RB, Kramer-Fox R, O'Ranghlin J (1989): Two dimensional aortic root dimensions in normal children and adults. Am J Cardiol 64:507-512. Roman MJ, Rosen SS, Kramer-Fox R, Devereux RB (1993): The prognostic significance of the pattern of aortic root dilatation in the Marfan syndrome. J Am Coll Cardiol 22:1470-1476. Royce PM, Steinmann B, Vogel A, Steinhorst U, Kohlschuetter A (1990): Brittle cornea syndrome: A heritable connective tissue disorder distinct from Ehlers-Danlos syndrome type VI and fragilitas oculi, with spontaneous perforations of the eye, blue sclerae, red hair and normal collagen lysyl hydroxylation. Eur J Pediatr 149:465-469. Sacheti A, Szemere J, Bernstein B, Tafas T, Schechter N, Tsipouras P (1997): Chronic pain is a manifestation of the Ehlers-Danlos syndrome. J Pain Symptom Managem in press. Sorokin Y, Johnson MP, Rogowski N, Richardson DA, Evans MI (1994): Obstetric and gynecologic dysfunction in the Ehlers-Danlos syndrome.
J Reprod Med 39:281-284. Steinmann B, Superti-Furga A, Joller-Jemelka HJ, Cetta G, Byers PH (1989): Ehlers- Danlos syndrome type IV: a subset of patients distinguished by low serum levels of the amino-terminal propeptide of type III procollagen. Am J Med Genet 34:68-71. Steinmann B, Royce PM, Superti-Furga A (1993): The Ehlers-Danlos syndrome. In: Connective Tissue and its Heritable Disorders: Molecular, Genetic and Medical Aspects. (Royce PM, and Steinmann B. eds): 351-407, Wiley-Liss, New York . Steinmann B, Eyre DR, Shao P (1995): Urinary pyridinoline cross-links in Ehlers- Danlos syndrome type VI. Am J Hum Genet 57:1505-1508. Stewart RE, Hollister DW, Rimoin DL (1977): A new variant of Ehlers-Danlos syndrome: An autosomal dominant disorder of fragile skin, abnormal scarring and generalized periodontitis. Birth Defects: Original Article Series: XIII, 3B:85- 93. Vogel A, Holbrook KA, Steinmann B, Gitzelmann R, Byers PH (1979): Abnormal collagen fibril structure in the gravis form (type I) of the Ehlers-Danlos syndrome. Lab Invest 40:201-206. Wenstrup RJ, Murad S, Pinnell SR (1989): Ehlers-Danlos syndrome type VI: Clinical manifestations of collagen lysyl hydroxylase deficiency. J Pediatr 115:405-409. Wolf F, Smythe HA, Yunus MB, Bennett RM, Bombardier C, Goldenberg DL, Tugwell P, Campbell SM, Abeles M, Clark P, Fam AG, Farber SJ, Fiechtner JJ, Franklin CM, Gatter RA, Hamaty D, Lessard J, Lichtbroun AS, Masi AT, McCain GA, Reynolds WJ, Romano TJ, Russell IJ, Sheon (1990): The American College of Rheumatology 1990 criteria for the classification of fibromyalgia. Arthritis and Rheumatism